To estimate the influence of the applied approximations on the
description of protein structure and dynamics, we have extracted
the following observables from the long time simulations.
As a measure for the temporal evolution of protein structure
we have chosen the rms deviations
![]() of the intermediate configurations from the
initial structure.
If
of the intermediate configurations from the
initial structure.
If ![]() are the atomic positions obtained after the
equilibration phase, the rms
deviations of the set
are the atomic positions obtained after the
equilibration phase, the rms
deviations of the set ![]() of heavy backbone atoms
(
of heavy backbone atoms
(![]() ) and of
the corresponding set
) and of
the corresponding set ![]() of side chain atoms
(
of side chain atoms
(![]() ) are given by
) are given by
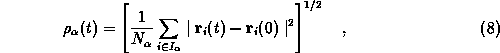
where ![]() denotes the number of atoms in the set
denotes the number of atoms in the set
![]() .
.
To estimate approximation effects on protein dynamics we monitor
correlations between the motions of atoms i and
j by the
normalized covariance matrix
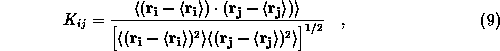
where the averages are taken over trajectory sections specified
further below.
Subsequently we compare covariance matrices ![]() and
and
![]() from simulations
A and
B, respectively, both by
a graphical method and by a single numerical observable.
The latter is the rms difference of the cross correlations,
i.e.,
from simulations
A and
B, respectively, both by
a graphical method and by a single numerical observable.
The latter is the rms difference of the cross correlations,
i.e.,
![]()
whereas the former is an overlay of certain contour plots pertaining
to the ![]() .
These contour plots visualize histograms representing the frequencies
of values
.
These contour plots visualize histograms representing the frequencies
of values ![]() as functions of atomic distances
as functions of atomic distances
![]() and will be called covariance plots.
and will be called covariance plots.
As a technical point we would like to note that comparisons of the type sketched above require, that the trajectories refer to one and the same conformational substate of the protein considered. The reason is, that dynamical correlations may vary from substate to substate, such that differences of these correlations, which are caused by the application of different algorithms, may be obscured.