In order to solve the classical equations of motion numerically,
and, thus,
to obtain the motion of all atoms the
forces acting on every atom have to be computed at each integration
step. The forces are
derived from an energy function which defines the molecular
model [1,2,3]. Besides other
important contributions (which we shall not discuss here) this
function contains the Coulomb sum
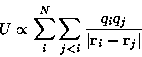
A very simple -- and in fact quite widely used -- approximation completely neglects long range electrostatic interactions beyond a certain cut-off distance [43] of typically 8-15Å. For systems which are significantly larger than this cut-off distance the computation of the remaining Coulomb interactions then scales with N instead of N2. However, such truncation leads to serious artifacts concerning the description of the structure and dynamics of proteins [44,24,45], and more accurate methods which include the long range interactions should be preferred. Multipole methods and multiple-time-step methods are well established and widely used for this purpose. We briefly sketch both methods and subsequently show how their combination allows highly efficient simulations.