Multipole methods approximate the long-range forces originating from a group of point charges by truncated multipole expansions of their electrostatic potential. Using a hierarchy of grids for subdivision of space, nested at multiple scales, and a corresponding hierarchical organization of charge groups and multipole expansions [33] a computational complexity of O(N log N) can be achieved. By additionally using a hierarchy of local Taylor expansions for the evaluation of the electrostatic potential in the vicinity of a group of particles Greengard and Rokhlin have constructed the so-called fast multipole method (FMM) that even scales with O(N) for large systems [34,35].
For MD simulations of biomolecules the FMM-type grouping of charges, defined by a fixed and regular subdivision of space, requires multipole expansions of rather high order (more than 6 terms of the expansion) as to achieve sufficient numerical accuracy [34]. If, instead, as shown in Figure 1, the charge grouping is adapted to specific structural and dynamical properties of the simulated biomolecules, the multipole expansions can be truncated at quite low orders, e.g., after the second order, while maintaining sufficient accuracy [36,37,38].
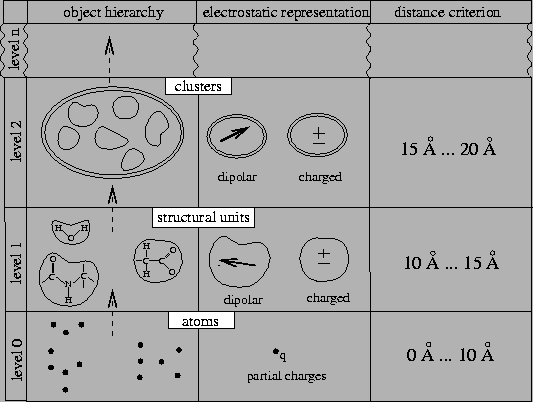 |
In the FAMUSAMM framework, e.g., we have grouped locally
stable groups of typically three to ten covalently bound atoms
into so-called structural units (level 1 in
Fig. 1). By construction, these structural units
either carry integer elementary charges or are uncharged, but dipolar.
Test simulations show that for distances
![]() Å already the lowest
non-vanishing multipole moments of these structural units provide
a sufficiently accurate description of the electrostatic forces within
biomolecules with an error below 2%. The objects of the next
hierarchy level (level 2 in the figure) are formed by grouping
structural units into clusters. For interaction distances
Å already the lowest
non-vanishing multipole moments of these structural units provide
a sufficiently accurate description of the electrostatic forces within
biomolecules with an error below 2%. The objects of the next
hierarchy level (level 2 in the figure) are formed by grouping
structural units into clusters. For interaction distances
![]() Å the electrostatic potential of
those objects, again, can be approximated by their lowest
multipole moment. Extending this scheme to higher hierarchy
levels, such a structure adapted multipole method (SAMM)
provides a substantial speed-up for MD simulations as compared
to the conventional, grid-based
methods [38,46].
Å the electrostatic potential of
those objects, again, can be approximated by their lowest
multipole moment. Extending this scheme to higher hierarchy
levels, such a structure adapted multipole method (SAMM)
provides a substantial speed-up for MD simulations as compared
to the conventional, grid-based
methods [38,46].
The performance of this first version of SAMM [36] can be further enhanced by additionally utilizing FMM-strategies [34,38]. Here, in the vicinity of a given object (e.g., a structural unit or a cluster) the electrostatic potential originating from distant charge distributions is approximated by a local Taylor expansion. Specifically, the basic tasks involved in the FMM aspect of SAMM are:
In the next section we will illustrate how to further speed up the SAMM method by introducing multiple-time-stepping.