As sketched in the right part of the figure, forces between distant atoms generally exhibit slower fluctuations than forces between close atoms. Therefore, without significant loss of accuracy, the more slowly fluctuating forces may be computed using larger integration step sizes. As shown in the left part of the figure, the required classification of forces can be implemented, e.g., by grouping atom pairs into distance classes. The slowly fluctuating forces arising from outer distance classes may then be evaluated less frequently (filled squares) than the fast ones and, instead, are extrapolated (open squares) from previously computed forces at the time steps in between.
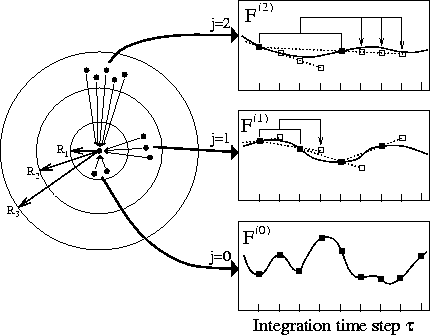 |
This hierarchical extrapolation procedure can save a significant amount of computer time as it avoids a large fraction of the most time consuming step, namely the exact evaluation of long range interactions. Here, computational speed is gained at the cost of an increased demand for memory, e.g., for each atom and each distance class two previously computed forces have to be kept in memory.
In the framework of the fast structure adapted multipole method the memory demand could be drastically reduced. This was achieved by applying the multiple-time-step scheme (we used the so-called DC-1d scheme [24]) to the interactions between charge groups (structural units and clusters) rather than to the forces acting on individual atoms. In the following we give a short description of this tight and efficient combination. We termed the resulting algorithm FAMUSAMM (multiple-time-step structure-adapted multipole method) [46,47].
A detailed analysis of fast SAMM has shown [47]
that the most time consuming tasks are task 2 and task 4
described above.
In task 2 for each hierarchy level (except for level 0)
a local Taylor expansion is calculated for each object.
Note that here we refer to expansions which comprise
only contributions from objects of the same hierarchy level
which, in addition, fulfill the distance criterion
given in Fig. 1. From each of these local
expansions, approximated electrostatic
forces ![]() acting on the atoms contained in
the associated object could be computed and, in analogy to
the exact forces
acting on the atoms contained in
the associated object could be computed and, in analogy to
the exact forces ![]() used in the multiple time step
scheme described above (see Fig. 2), the
multipole-derived forces
used in the multiple time step
scheme described above (see Fig. 2), the
multipole-derived forces ![]() could be
extrapolated by multiple time stepping.
We further improved that obvious scheme, however, in that we applied
multiple time step extrapolations
to the coefficients of the local Taylor expansions instead.
That strategy reduces memory requirements by a significant factor
without loss of accuracy, since the number of local Taylor coefficients
that have to be kept for the extrapolation is
smaller than the number of forces acting on all atoms of the respective
object.
could be
extrapolated by multiple time stepping.
We further improved that obvious scheme, however, in that we applied
multiple time step extrapolations
to the coefficients of the local Taylor expansions instead.
That strategy reduces memory requirements by a significant factor
without loss of accuracy, since the number of local Taylor coefficients
that have to be kept for the extrapolation is
smaller than the number of forces acting on all atoms of the respective
object.
Additionally, to optimize task 4, we applied a conventional,
atom pair interaction based multiple-time-step scheme to
the force computation within the innermost distance class.
Here, for atom pairs closer than ![]() Å, the Coulomb sum is
calculated every step, and for all other atom pairs the Coulomb
sum is extrapolated every second step from previously
explicitly calculated forces.
Å, the Coulomb sum is
calculated every step, and for all other atom pairs the Coulomb
sum is extrapolated every second step from previously
explicitly calculated forces.
This completes the outline of FAMUSAMM. The algorithm has been implemented in the MD simulation program EGO_VIII [48] in a sequential and a parallelized version; the latter has been implemented and tested on a number of distributed memory parallel computers, e.g., IBM SP2, Cray T3E, Parsytec CC and ethernet-linked workstation clusters running PVM or MPI.