The previous application -- in accord with most MD studies -- illustrates the urgent need to further push the limits of MD simulations set by todays computer technology in order to bridge time scale gaps between theory and either experiments or biochemical processes. The latter often involve conformational motions of proteins, which typically occur at the microsecond to millisecond range. Prominent examples for functionally relevant conformational motions are the opening and closing of ion channels or, as proposed by Griffith [55] and Prusiner [56], pathogenic conformational transitions in prion proteins, the putative agents of mad cow and Creutzfeldt-Jacob diseases. Conformational motions often involve a complex and concerted rearrangement of many atoms in a protein from its initial state into a new conformation. These rearrangements, called conformational transitions, exhibit a multi-rate behaviour, which is is captured by the concept of ``hierarchical conformational substates'' introduced by Hans Frauenfelder [57]. According to that concept the free energy landscape of a protein exhibits a large number of nearly isoenergetic minima corresponding to the conformational substates, which are separated by barriers of different height [58].
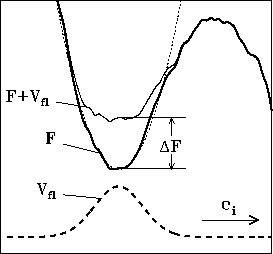 |
Figure 8 shows a one-dimensional sketch of a small fraction of that energy landscape (bold line) including one conformational substate (minimum) as well as, to the right, one out of the typically huge number of barriers separating this local minimum from other ones. Keeping this picture in mind the conformational dynamics of a protein can be characterized as ``jumps'' between these local minima. At the MD time scale below nanoseconds only very low barriers can be overcome, so that the studied protein remains in or close to its initial conformational substate and no predictions of slower conformational transitions can be made.
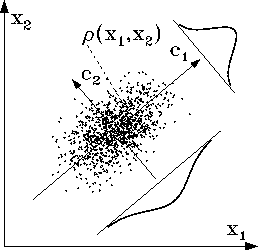 |
In order to make such predictions possible, we have developed the
conformational flooding (CF) method, which accelerates
conformational transitions [59] and thereby brings them
into the scope of MD simulations (``flooding simulations''). The method
is a generalization of the ``local elevation method'' [60]
in that it rests
on a quasi harmonic model for the free energy landscape in the vicinity of the
minimum representing the initial (known) conformational state. This model is
derived from an ensemble of structures generated by a conventional MD
simulation as will be described below and is shown in Fig. 9.
From that model a ``flooding potential'' ![]() is constructed (dashed
line in Fig. 8), which, when subsequently
included into the potential energy
function of the system, raises the minimum under consideration (thin line
in Fig. 8) and thereby lowers the surrounding free energy
barriers by an amount
is constructed (dashed
line in Fig. 8), which, when subsequently
included into the potential energy
function of the system, raises the minimum under consideration (thin line
in Fig. 8) and thereby lowers the surrounding free energy
barriers by an amount ![]() without severely modifying the barriers
themselves. As a result, transitions over these barriers are accelerated
by approximately the Boltzmann factor
without severely modifying the barriers
themselves. As a result, transitions over these barriers are accelerated
by approximately the Boltzmann factor ![]() .
In detail, the following steps are necessary to perform a CF simulation:
.
In detail, the following steps are necessary to perform a CF simulation:
Step 1: A short conventional MD simulation (typically
extending over a few 100ps)
is performed to generate
an ensemble of protein structures ![]() (each described by N atomic positions),
which characterizes the initial conformational substate.
The 2-dimensional sketch in Fig. 9 shows such an ensemble
as a cloud of dots, each dot x representing one ``snapshot'' of the
protein.
(each described by N atomic positions),
which characterizes the initial conformational substate.
The 2-dimensional sketch in Fig. 9 shows such an ensemble
as a cloud of dots, each dot x representing one ``snapshot'' of the
protein.
Step 2: This ensemble
is subjected to a ``principal component analysis'' (PCA) [61] by
diagonalizing the covariance matrix
![]() ,
,
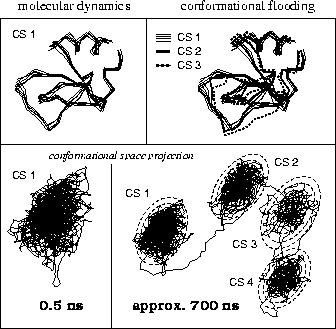 |
As a sample application we describe simulations suggesting possible conformational transitions of the protein BPTI (Bovine Pancreatic Trypsin Inhibitor) at a time scale of several 100 nanoseconds (see Fig. 10). First we carried out a conventional MD simulation of 500 ps duration (no explicit solvent included), during which the protein remained in its initial conformational substate CS 1. The upper left part of the figure shows several snapshots of the backbone taken from that simulation; the lower left shows a projection of the 500 ps trajectory onto the two conformational coordinates with largest eigenvalues (corresponding to Fig. 9). From that ensemble we constructed a flooding potential as described above (dashed contour lines, superimposed to the CS 1-trajectory, bottom right). The flooding potential was subsequently switched on and rapidly induced a conformational transition (to the right in the figure) into another energy minimum, CS 2. After switching off the flooding potential the new conformational state of the protein remained stable, indicating that, indeed, the new minimum is separated from CS 1 by a large energy barrier. Using multi-dimensional transition state theory [59] we could estimate that in an conventional (i.e., unperturbed) MD simulation that conformational transition would have been observed only after several hundred nanoseconds. As shown in Fig. 10, the CF method can be applied iteratively to systematically search for further conformational substates, CS 3, CS 4 etc. The upper right part of the figure shows the backbone configuration of BPTI corresponding to the new substates.
MD simulations are valuable tools if one wants to gain detailed insight into fast dynamical processes of proteins and other biological macromolecules at atomic resolution. But since conventional MD simulations are confined to the study of very fast processes, conformational flooding represents a complementary and powerful tool to predict and understand also slow conformational motions. Another obvious application is an enhanced refinement of Xray- or NMR-structures.