Here we want to document that FAMUSAMM actually provides an enhanced computational efficiency both as compared to SAMM as well as to the reference method with its exact evaluation of the Coulomb sum. For that purpose we first consider how the computational effort of FAMUSAMM scales with system size.
For a quantitative performance assessment we have carried out a series of
test simulations on systems of varying size using the sequential version
of our MD program EGO_VIII.
The chosen system sizes covered the range from 500 to
40,000 atoms.
All simulations were executed on a
DEC-ALPHA 3300L (175 MHz) workstation equipped with
96 MB RAM .
Figure 8 shows how the average computation time required for one
MD integration
step scales with system size. The choice of an average computation time for
performance measurements is necessary, since in FAMUSAMM the time
required for the force calculation varies from time step to time step
(cf. Figure 4).
.
Figure 8 shows how the average computation time required for one
MD integration
step scales with system size. The choice of an average computation time for
performance measurements is necessary, since in FAMUSAMM the time
required for the force calculation varies from time step to time step
(cf. Figure 4).
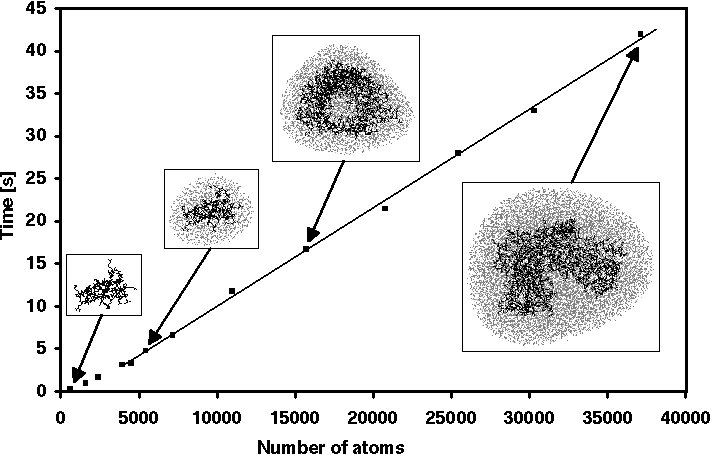
Figure: Average computation time for one step using EGO_VIII on a DEC-Alpha 3300L workstation
(175 MHz) for simulation systems of varying size.
Figure 8 clearly shows that for systems comprising more than about 1,000 atoms FAMUSAMM achieves linear scaling of computational effort with size. The quadratic increase observed for very small systems is in line with the fact, that in the innermost distance class (H=0 in Figure 4) forces are calculated by use of the Coulomb sum with its intrinsically quadratic scaling behavior. Thus, the theoretically expected linear scaling of FAMUSAMM for large systems is actually obtained in our implementation.
For performance comparisons with other methods consideration of system size is essential. As compared to its parent method SAMM, our new algorithm achieves a speed-up by a factor of two for small systems containing less than about 5,000 atoms. This gain of efficiency is entirely due to our multiple-time-step extrapolation of forces in the innermost SAMM distance class H=0 [see subsection c) in section ``Combination of SAMM with a multiple-time-step method'' as well as Figure 4]. Note that SAMM had been previously demonstrated to perform for systems up to that size as efficiently as the conventional cutoff methods [26]. Thus, FAMUSAMM beats the cutoff method in this regime also concerning efficiency.
For large systems comprising 36,000 atoms FAMUSAMM performs four times faster than SAMM and as fast as cutoff. Here, the speed-up with respect to SAMM is essentially achieved by the multiple-time-step extrapolation of local Taylor expansions in the outer distance classes, and FAMUSAMM executes by a factor of 60 faster than the reference method based on the evaluation of the Coulomb sum.